
BIOLOGIA |
Muerte celular programada: Un proceso clave en fisiología y patología. |
RESUMEN [ABSTRACT]
La idea de que la muerte, incluso cuando ocurre a nivel de las células que componen un organismo vivo, es un proceso lamentable y frecuentemente accidental ha cambiado en los últimos años. Estudios de Biología del Desarrollo en diversos organismos modelo han demostrado que la muerte celular es un proceso que se produce naturalmente en el desarrollo normal de organismos pluricelulares, desde gusanos hasta la especie humana. El análisis genético, realizado inicialmente en Caenorhabditis elegans, ha demostrado que la muerte celular es un proceso regulado a nivel genético: las células expresan los componentes moleculares que les van a permitir "suicidarse" dependiendo de un balance de señales procedentes del medio ambiente celular. La caracterización de este complejo mecanismo ha cambiado profundamente la comprensión de numerosas patologías humanas. Por ejemplo, el cáncer frecuentemente se desarrolla porque disminuye la capacidad de muerte celular, mientras que un exceso de muerte celular desencadena patologías degenerativas. En este artículo, además, presentamos los mecanismos de la muerte celular programada, los cuales constituyen, en nuestra opinión, el aspecto biológico esencial, basándonos principalmente en las aportaciones provenientes de estudios sobre el desarrollo del sistema nervioso.
Muerte celular programada.
La muerte celular programada es un proceso de autodestrucción celular controlada que permite al organismo su correcta morfogénesis, así como su renovación y la eliminación de las células que amenacen su supervivencia. Esta muerte es de vital importancia, tanto durante el desarrollo embrionario como durante la vida adulta (Glücksmann, 1951; Kerr et al., 1972; Jacobson et al., 1997; Raff, 1998).
Los sistemas modelo de desarrollo embrionario han proporcionado buena parte de las observaciones disponibles sobre muerte celular programada. Uno de los ejemplos más visibles del resultado de la muerte celular programada es la morfogénesis de los dedos, que se produce por eliminación de las áreas interdigitales. La muerte celular programada origina que los humanos tengamos 5 dedos en cada extremidad. Su ausencia en los patos, por ejemplo, les hace conservar su característica pata palmeada. Otro ejemplo clásico es la muerte neuronal. Durante el desarrollo se producen neuronas en exceso, lo que permite posteriormente un refinamiento de la inervación al morir aquellas neuronas menos capacitadas, en un a modo de selección darwiniana a nivel celular. El sistema inmune también proporciona muchos de los ejemplo clásicos de muerte celular programada. Tanto en la selección del repertorio de linfocitos que han de defender al organismo, produciéndose la eliminación de aquellos que reconocen antígenos propios, como en la eliminación de células infectadas o tumorales por citolisis se requiere un correcto funcionamiento del mecanismo de muerte celular programada.
Necrosis, apoptosis, muerte celular programada.
La acumulación de observaciones, provenientes en su mayor parte de sistemas modelo de desarrollo embrionario, pusieron de manifiesto algunas peculiaridades morfológicas de las células que morían en condiciones fisiológicas (Kerr et al., 1972). De esta forma se acuñó el término apoptosis, por contraposición a la necrosis de las situaciones patológicas. Los términos necrosis y apoptosis hacen principalmente referencia a los aspectos morfológicos de las células que mueren. Una célula necrótica se hincha, explota y libera su contenido citoplásmico, lo que produce una respuesta inflamatoria al atraer células del sistema inmune. Originalmente se pensaba que todas las muertes celulares, o al menos una gran mayoría, presentaban esta morfología. Sin embargo, esta muerte está restringida a situaciones "accidentales" o agudas: heridas, infecciones, el daño inicial en infartos, etc... Su asociación con la respuesta inflamatoria la hacen fácilmente detectable. Una célula apoptótica, por el contrario, va reduciendo paulatinamente su volumen y perdiendo primero porciones de citoplasma rodeado de membrana (Figura 1). Más adelante su cromatina también se va a fraccionar. Los cuerpos picnóticos, como se llaman dichos fragmentos, son engullidos por células vecinas y pueden desaparecen, en tan sólo una hora o poco más, sin dejar rastro ni inducir una respuesta inflamatoria. Es una muerte que ha permanecido desconocida por su discreción, aunque ahora se sabe que es la mayoritaria, tanto en procesos fisiológicos como patológicos.
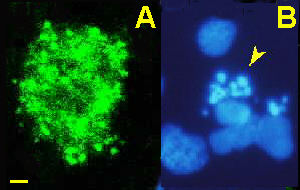
Figura 1. Apariencia de las células apoptóticas. Células neuroepiteliales de la retina de embrión de pollo teñidas con un marcador de caspasa 3 activada (A), lo que permite visualizar los eventos iniciales con la pérdida de vesículas de membrana citoplásmica. La tinción de la cromatina (B) permite visualizar los eventos más tardíos de apoptosis nuclear por fragmentación del núcleo en cuerpos picnóticos (flecha)
El término muerte celular programada, más preciso desde el punto de vista del mecanismo, responde a estudios más recientes que demostraron la existencia de una maquinaria intracelular de muerte cuyos componentes, codificados genéticamente, se expresan en todas las células nucleadas del organismo (Weil et al., 1996). Es decir, existe un "programa" que controla el mecanismo de muerte celular. Es fácil entender la necesidad de una precisa y estricta regulación de un proceso irreversible como la muerte celular. La exposición, incluso simplificada, de sus mecanismos básicos tiene un grado de complejidad bastante por encima del nivel divulgativo. Por ello lo hemos recogido como un Apéndice aparte del texto principal. La muerte celular programada normalmente tiene lugar por apoptosis, aunque existen bastantes excepciones que requieren aún un estudio detallado del mecanismo subyacente..
Muerte celular en fisiología normal y en situaciones patológicas.
Durante décadas, aunque se conocían ejemplos como los citados anteriormente del desarrollo embrionario, se seguía considerando a la muerte celular por apoptosis un proceso singular, aunque fisiológico, de ciertos sistemas específicos. Sin embargo, según se iba observando muerte en nuevos tipos celulares y procesos del desarrollo, fue cambiando la visión hacia considerar que la muerte celular programada es un proceso mucho más general de lo que en un principio se pensaba (Raff, 1992). En un claro ejemplo de cómo estudios en un principio de carácter básico acaban teniendo, inesperadamente, una enorme incidencia en Medicina, el estudio de la muerte celular programada se considera ya esencial en numerosas patologías (Thompson, 1995; Naik et al., 1996; Stambolic et al., 1999; Lockshin et al., 2000). La temida progresión tumoral que tantas vidas cuesta no parece depender sólo de la capacidad proliferativa e invasiva, sino también de la pérdida de la regulación de la muerte celular por parte de las células tumorales: dejan de responder tanto a sus controles internos, que las llevarían a "suicidarse", como a algunas de las señales que con el mismo fin les envía el sistema inmune. Numerosos virus que infectan nuestras células codifican para moléculas inhibidoras del programa de apoptosis. Así evitan que la célula muera, de nuevo por indicaciones de las células del sistema inmune, y así disponen de mayor tiempo para multiplicarse. La muerte celular programada también es esencial, en correcto equilibrio con la proliferación, para la renovación de los tejidos que constituyen el organismo. Es fácil comprender que una aceleración del proceso de muerte va a originar procesos degenerativos. El sistema nervioso es especialmente sensible a estos procesos por su reducida o ausente capacidad proliferativa. Factores hereditarios o ambientales van a desregular el proceso de muerte celular programada y producir enfermedades tales como la enfermedad de Alzheimer, diferentes tipos de esclerosis y una larga lista de males de consecuencias personales y sociales muy dolorosas.
El conocimiento de los mecanismos moleculares (Ver Apéndice y Figura 2) que llevan a una célula a su muerte programada en situaciones fisiológicas puede ayudar a remediar la pérdida irreparable de células en ciertas situaciones patológicas y disminuir así las secuelas de ciertas enfermedades. Asimismo, conociendo en detalle el mecanismo de muerte celular, puede ser mejorada la terapia de inducción activa de muerte celular en poblaciones de células tumorales, efecto principal de la radioterapia y quimioterapia actuales, en búsqueda de tratamientos con menores efectos secundarios.
Bibliografía
Glüksmann, A. (1951). Cell death in normal vertebrate ontogeny. Biol. Rev. 26, 59-86.
Jacobson M.D., Weil, M. & Raff, M.C. (1997). Programmed cell death in animal development. Cell 88, 347-354.
Kerr, J.F.R., Wyllie, A.H. & Currie, A.R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implication in tissue kinetics. Br. J. Cancer 26, 239-257.
Lockshin, R.A., Osborne, B. & Zakeri, Z. (2000). Cell death in the third millenium. Cell Death Diff. 7, 2-7.
Naik, P., Karrim, J. & Hanahan, D. (1996). The rise and fall of apoptosis during multistage tumorigenesis:down-modulation contributes to tumor progression from angiogenic progenitors. Genes Dev. 10, 2105-2116.
Raff, M.C. (1992). Social controls on cell survival and cell death. Nature 356, 397-400.
Raff, M.C. (1998). Cell suicide for beginners. Nature 396, 119-122.
Stambolic, V., Mak, T.W. & Woodgett, J.R. (1999). Modulation of cellular apoptotic potential: contribution to oncogenesis. Oncogene 18, 6094-6103.
Thompson, C.G. (1995). Apoptosis in the pathogenesis and treatment of disease. Science 267, 1456-1462.
Weil, M., Jacobson, M.D., Coles, H.S.R., Davies, T.J., Gardener, R.L., Raff, K.D. & Raff, M.C. (1996). Constitutive expression of the machinery for programmed cell death. J.Cell Biol. 133, 1053-1059.
Apéndice
Mecanismo molecular de la muerte celular programada.
Muchos de estos mecanismos se han conservado a lo largo de la escala evolutiva desde nemátodos hasta mamíferos (Vaux et al., 1992; Hengartner & Horvizt, 1994). Si se superan los mecanismos de control intra y extracelulares, independientemente de la naturaleza del estímulo inductor de muerte, el proceso siempre resulta en la activación de una cascada de proteasas que desmantelan la célula de forma rápida y controlada, sin causar daños en el entorno celular (Figura 2).
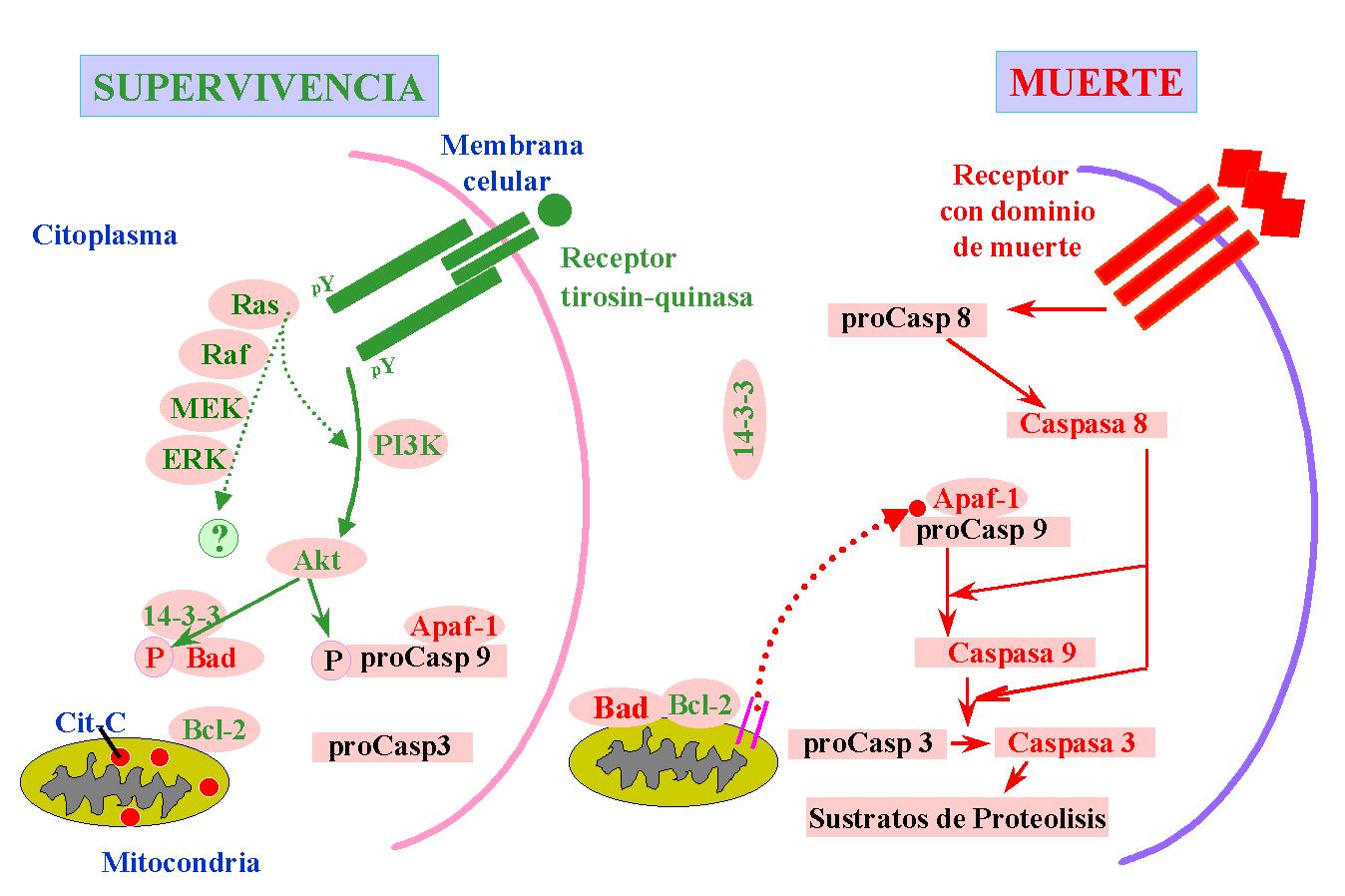
Figura 2. Esquema representativo del mecanismo central de apoptosis. En verde se representan las moléculas anti-apoptóticas y en rojo las pro-apoptóticas.
Efectores intracelulares
El mecanismo central de la maquinaria de muerte en cualquier célula está constituido por una familia de proteasas, a las que se han denominado "caspasas" (Alnemri et al., 1996), por ser Cisteín-proteASAS que hidrolizan a su sustrato junto a un residuo de ácido ASPártico. Las caspasas responden al estímulo apoptótico mediante activación de una cascada intracelular proteolítica que ocasiona la activación o inactivación de diferentes sustratos celulares y provoca la muerte celular. La primera caspasa que se describió fue la caspasa-1 o ICE (del Inglés, Interleukin-1b -Converting Enzyme), homóloga en mamíferos de la proteína CED-3 de Caenorhabditis elegans (Thornberry et al.,1992; Yuan et al., 1993), producto del gen ced-3, necesario para la muerte celular programada en este nemátodo (Ellis et al., 1991; Xue & Horvitz, 1995). Se conocen actualmente 13 caspasas en mamíferos (Thornberry & Lazebnik, 1998). De ellas, 6 se relacionan preferentemente con procesos inflamatorios y no con procesos de muerte celular, aunque todas comparten semejanzas en cuanto a secuencia proteica, estructura y especificidad de sustrato (Nicholson & Thornberry, 1997).
Las caspasas son sintetizadas como proenzimas inactivas, denominados procaspasas, que, una vez activados por proteólisis limitada y asociación de subunidades (Stennicke & Salvesen, 1998), adquieren la actividad catalítica para proteolizar a su vez a sus sustratos tras un residuo de aspártico. Reconocen, sin embargo, diferentes motivos tetrapeptídicos, lo cual les confiere cierta especificidad de sustrato. Las caspasas inactivas pueden ser sustrato a su vez de las activas, de manera que unas caspasas activan a otras siguiendo un orden jerárquico y una actuación en cascada (Salvesen & Dixit, 1997).
Las caspasas que participan en apoptosis pueden dividirse en dos grandes grupos: caspasas iniciadoras (8 y 9, y quizás 2 y 10), que se activan en respuesta a señales apoptóticas y activan a las caspasas efectoras (3, 6 y 7), las cuales proteolizan sustratos celulares provocando la desorganización de la célula y los cambios morfológicos típicos de la apoptosis (revisado en Thornberry & Lazebnik, 1998). Además, parece existir especificidad de tejido en cuanto a la caspasa requerida para el proceso apoptótico. Los ratones mutantes nulos para la caspasa-3 mueren al nacer presentando un número superior al normal de células en el cerebro, pero ninguna anomalía en el resto de sus órganos (Kuida et al., 1996). Además de la caspasa-3, también pueden estar implicadas en procesos de muerte neuronal otras caspasas. La caspasa-2 se expresa a altos niveles en el cerebro del ratón embrionario, pero no en el adulto (Kumar et al., 1994), y su mutante nulo presenta alteración en la muerte de las neuronas simpáticas en ausencia de NGF (del Inglés, Nerve Growth Factor) (Bergeron et al., 1998). La caspasa-1 se ha implicado en algunos procesos de muerte neuronal (Troy et al., 1996; Tanaka et al., 1998), aunque el ratón mutante nulo para este gen no presenta ninguna alteración en el desarrollo del cerebro (Kuida et al., 1995).
Se conocen unos 40 sustratos celulares para las caspasas (Cryns & Yuan., 1998). Entre estos se encuentran proteínas que inhiben muerte celular, como Bcl-2 (Cheng et al, 1997), proteínas que participan en la reparación del DNA o en la organización de la cromatina (Kaufmann et al., 1993; Casciola-Rosen et al., 1996), y proteínas implicadas en la regulación del citoesqueleto (Rudel & Bokoch, 1997). En la muerte celular del sistema nervioso puede participar otra proteasa, la calpaína, cisteín proteasa dependiente de Ca2+ (Squier et al., 1994). En ocasiones la apoptosis neuronal requiere participación conjunta de calpaína y caspasas (Jordan et al., 1997).
Reguladores intracelulares
Los principales reguladores intracelulares de la muerte celular programada son la familia de proteínas relacionadas con Bcl-2, proteína de mamíferos homóloga estructural y funcionalmente a la proteína CED-9, esencial para la prevención de la muerte celular en C. elegans (Hengartner & Horvitz., 1994). La familia de Bcl-2 en mamíferos está constituida hasta el momento por 15 miembros (Chao & Korsmeyer, 1998; Adams & Cory, 1998) y todos ellos presentan al menos uno de los cuatro dominios conservados de homología a Bcl-2 (BH1 a BH4). Algunos miembros de la familia inhiben la muerte celular (Bcl-2, Bcl-XL) y otros la activan (Bax, Bad, Bid). Puesto que ambos tipos pueden heterodimerizar, sus concentraciones relativas pueden regular el balance entre muerte y supervivencia (Oltvai et al., 1993). Muchas de estas proteínas presentan un dominio c-terminal hidrofóbico que les permite anclarse a la cara citoplasmática de las membranas de la mitocondria, retículo endoplásmico y envuelta nuclear, de forma que pueden registrar daños en dichos orgánulos (revisado en Green & Reed, 1998). El siguiente nivel de regulación se produce en la interacción de CED-4 en C. elegans (Chinnaiyan et al., 1997; Pan et al., 1998) y su homólogo Apaf-1 en mamíferos con algunas de las caspasas iniciadoras. Bcl-XL y Bcl-2 impiden las activación del complejo Apaf-1/procaspasa-9 (Hu et al., 1998), además de impedir directa o indirectamente la liberación de Citocromo C de la mitocondria, lo que a su vez parece ser responsable de un cambio conformacional en Apaf-1 que le permite procesar a la procaspasa-9 (Zou et al., 1997; Li at al., 1997; Green & Reed, 1998). Aún no está claro si la liberación de Citocromo C forma parte del mecanismo inicial de activación de las caspasas, o es un paso de amplificación de la respuesta apoptótica (Adams & Cory, 1998).
Bcl-2, Bcl-XL y Bax participan en muerte celular en el sistema nervioso (revisado en Pettmann & Henderson, 1998). Bcl-XL está ampliamente expresado en sistema nervioso (González-García et al., 1995), y los ratones mutantes nulos para este gen, que mueren durante el desarrollo embrionario, presentan muerte masiva de neuronas postmitóticas inmaduras en cerebro, médula espinal y ganglios de la raíz dorsal (Motoyama et al., 1995). Los ratones mutantes nulos para Bcl-2 completan su desarrollo embrionario, pero mueren en torno a los 30 días postnatales, mostrando pérdida progresiva de motoneuronas faciales y sensoriales (Michaelidis et al., 1996; Piñón et al., 1997). Los mutantes que sobreexpresan Bcl-XL ó Bcl-2 presentan un incremento en el número de neuronas sensoriales, y una mayor resistencia a morir frente a estímulos del programa de muerte (Martinou et al., 1994; Parsadanian et al., 1998). Los mutantes nulos para Bax se desarrollan normalmente, pero presentan incremento en el número de motoneuronas faciales, que son más resistentes a morir frente a axotomía neonatal (Deckwerth et al., 1996). Los ratones mutantes dobles nulos para Bcl-XL y Bax mostraron que Bax es el responsable de mucha de la muerte neuronal de los mutantes nulos para Bcl-XL (Shindler et al., 1997; Knudson & Korsmeyer, 1997).
Reguladores extracelulares
Las proteínas de la familia de Bcl-2 y las procaspasas, a su vez, son reguladas por señales extracelulares de supervivencia o de muerte (Figura 2). Diversas familias de neurotrofinas y factores de crecimiento forman parte de las moléculas señalizadoras de supervivencia en el sistema nervioso. Sus señales de supervivencia se pueden transducir por la vía intracelular que implica a la PI3K (del Inglés, PhosphatidylInositol-3-Kinase), (Kulik & Weber, 1998), y la consecuente activación de la proteín-quinasa B ó Akt (Yao & Cooper, 1995; Dudek et al., 1997; Franke et al., 1997), o por la vía que implica a las MAPK (del Inglés, Mitogen-Activated Protein Kinases), (Párrizas et al., 1997; Núñez & del Peso, 1998). La activación de Akt supone la fosforilación directa de Bad (Datta et al., 1997) que se mantiene unido a la proteína 14-3-3. Por el contrario, Bad no fosforilado se une a Bcl-XL (ó Bcl-2) impidiendo que ejerza su acción de supervivencia (Zha et al., 1996).
Existe, además, otra vía de inducción de muerte (Figura 2) que implica la señalización a través de los llamados receptores de muerte. Los mejor caracterizados son Fas y receptor de TNF (del Ingles, Tumor Necrosis Factor), a los que se unen el ligando de Fas y TNF respectivamente, y que participan en la apoptosis del sistema inmune (revisado en Ashkenazi & Dixit, 1998). Un caso sorprendente, que tiene lugar durante el desarrollo del sistema nervioso, es el de la activación por NGF del receptor de neurotrofinas p75, que contiene una secuencia con homología de dominio de muerte. Este receptor participa en la apoptosis inducida por NGF en retina de pollo (Frade et al., 1996), y es esencial para la muerte celular que afecta durante el desarrollo a las neuronas simpáticas (Bamji et al., 1998). La sobreexpresión de su dominio intracelular puede inducir la muerte de algunas neuronas in vivo (Majdan et al., 1997). La unión de NGF a p75 puede producir liberación de ceramida y activación de JNK (del Inglés, c-Jun N-terminal Kinase) en células gliales, rutas ambas implicadas en muerte celular programada (Carter et al., 1996; Casaccia-Bonnefil et al., 1996), pero se desconoce aún cómo induce la activación de procaspasas.
Bibliografía del Apéndice
Adams, J.M. & Cory, S. (1998). The Bcl-2 protein family: arbiters of cellular survival. Science 281, 1322-1326.
Alnemri, E.S., Livingston, D.J., Nicholson, D.W., Salvesen, G., Thornberry, N.A., Wong, W.W. & Yuan, J. (1996). Human ICE/CED-3 protease nomenclature. Cell 87, 171.
Ashkenazi, A. & Dixit, V.M. (1998). Death receptors: signalling and modulation. Science 281, 1305-1308.
Bamji, S.X., Majdan, M., Pozniak, D., Belliveau, DJ., Aloyz, R., Khon, J., Causing, C.G. & Miller, F.D. (1998). The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J. Cell Biol. 140, 911-923.
Bergeron, L., Perez, G.I., Mcdonald, G., Shi, L., Sun ,Y., Jurisicova, A., Varmuza, S., Latham, K.E.,, Flaws, J.A., Salter, J.C.M., Hara, H., Moskowitz, M.A., Li, E., Greenberg, A., Tilly, J.L. & Yuan, J. (1998). Defects in regulation of apoptosis in caspase-2 deficient mice. Genes Dev. 12, 1304-1314.
Carter, B.D., Kaltschmidt, C., Kaltschmidt, B., Offenhauser, N., Bohm-Matthaei, R., Baeuerle, P.A. & Barde, Y.A. (1996). Selective activation of NF-k B by nerve growth factor through the neurotrophin receptor p75. Science 272, 542-545.
Casaccia-Bonnefil, P., Carter, B.D., Dobrowsky, R.T. & Chao, M.V. (1996). Death of oligodendrocytes mediated by the interaction of nerve growth factor with is receptor p75. Nature 383, 716-719.
Casciola-Rosen, L., Nicholson, D.W., Chong, T., Rowan, K.R., Thornberry, N.A., Miller, D.K. & Rosen, A. (1996). Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J. Exp.Med. 183,1957-1964.
Chao, D.T. & Korsmeyer, S.J. (1998). Bcl-2 family: regulators of cell death. Annu. Revv Immunol. 16, 395-419.
Cheng, E.H. Kirsch, D.G., Clem, R.J., Ravi, R., Kastan, M.B., Bedi, A., Ueno, K. & Hardwick J.M. (1997). Bax-independent inhibition of apoptosis by Bcl-XL. Nature 379, 554-556.
Chinnaiyan, A.M., ORourke, K., Lane, B.R. & Dixit, V.M. (1997). Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cellular death. Science 275, 1122-1126.
Cryns, V. & Yuan, J. (1998). Proteases to die for. Genes Dev. 12, 1551-1570.
Datta, S.R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y. & Greenberg, M.E. (1997). Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell 91, 231-241.
Deckwerth, T.L., Elliot, J.L., Knudson, C.M., Johnson, E.M., Snider, W.D. & Korsmeyer, S.J. (1996). Bax is required for neuronal death after trophic factor deprivation and during development. Neuron 17, 401-411.
Dudek, H., Datta, S.R., Franke, T.F., Birnbaum, M.J., Yao, R., Cooper, G.M., Segal, R.A., Kaplan, D.R. & Greenberg, M.E. (1997). Regulation of neuronal survival by serine-threonine protein kinase Akt. Science 275, 661-665.
Ellis, R.E., Jacobson, D.M. & Horvitz, H.R. (1991). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics 129, 79-94.
Frade, J. M., Rodríguez-Tébar, A. & Barde, Y.-A. (1996). Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383, 166-168.
Franke, T.F., Kaplan, D.R. & Cantley, L.C. (1997). PI3K downstream AKTion blocks apoptosis. Cell 88, 435-437.
González-García, M. García, I., Ding, L., O'Shea, S., Boise, L. H., Thompson, C. B.& Núñez, G. (1995). bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death.Proc. Natl. Acad. Sci. USA 92, 4304-4308.
Green, D.R. & Reed, J.C. (1998). Mitochondria and apoptosis. Science 281, 1309-1312.
Hengartner, M.O. & Horvitz, H.R. (1994). C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian protooncogene bcl-2. Cell 76, 665-676.
Hu, Y., Benedict, M.A., Wu, D. Inohara, N. & Nuñez, G. (1998). Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase 9 activation. Proc. Natl. Acad. Sci. USA 95, 4386-4391.
Jordan, J., Galindo, M.F. & Miller, R.J. (1997). Role of calpain and interleukin 1b converting enzyme-life proteases in the b amyloid-induced death of rat hippocampal neurons in culture. J. Neurochem. 68, 1612-1621.
Kaufmann, S.H., Desnoyers, S., Ottavino, Y., Davidson, N.E. & Poirier, G.G. (1993). Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 53, 3976-3985.
Knudson, C.M. & Korsmeyer, S.J. (1997). Bcl-2 and Bax function independently to regulate cell death. Nature Genet. 16, 358-363.
Kuida, K., Lippke, J.A., Ku, G., Harding, M.W., Livingston, D.J., Su, M.S. &Flavell, R.A. (1995). Altered cytokine export and apoptosis in mice deficient in interleukin-1b converting enzyme. Science 267, 2000-2003.
Kuida, K., Zheng, T.S., Na, S., Kuan, C.Y., Yang, D., Karasuyama, H., Rakic, P. & Flavell, R.A. (1996). Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384, 368-372.
Kulik, G. & Weber, M.J. (1998). Akt-dependent and independent survival signalling pathways utilised by insulin-like growth factor I. Mol. Cell. Biol. 18, 6711-6718.
Kumar, S., Kinoshita, M., Noda, M., Copeland, N.G. & Jenkins, N.A. (1994). Induction of apoptosis by the mouse Nedd2 gene, which encodes a protein similar to the product of the Caenorhabditis elegans cell death gene ced-3 and the mammalian IL-1b -conveting enzyme. Genes Dev. 8, 1613-1626.
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S.M., Ahmad, M., Alnemri, E.S. & Wang, X. (1997).Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479-489.
Majdan, M., Lachance, C., Gloster, A., Aloyz, R., Zeindler, C., Bamji, S., Belliveau, D., Fawcet, J., Miller, F.D. & Barker, P.A. (1997). Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J. Neurosci. 17, 6988-6998.
Martinou, J.C., Dubois-Dauphin, M., Staple, J.K., Rdriguez, I., Frankowsky, H., Missotten, M., Albertini, P., Talabot, D., Catsicas, S., Pietra, C. & Huarte, J. (1994). Overexpression of bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischaemia. Neuron 13, 1017-1030.
Michaelidis, T.M., Sendtner, M., Cooper, J.D., Airksinen, M.S., Holtmann, B., Meyer, M., & Thoenen, H. (1996). Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron 17, 75-89.
Motoyama, N., Wang, F., Roth, K.A., Sawa, H., Nakayama, K., Negishi, I., Senju, S., Zhang, Q., Fuji, S. & Loh, D.Y. (1995). Massive cell death of immature hematopoietic cells and neurons in Bcl-X-deficient mice. Science 267, 1506-1510.
Nicholson, D.W. & Thornberry, N.A. (1997). Caspases: killer proteases. Trends Biochem. Sci. 22, 299-306.
Núñez, G. & del Peso, L. (1998). Linking extracellular survival signals and the apoptotic machinery. Curr. Op. Neurobiol. 8, 613-618.
Oltvai, Z.N., Milliman, C.L. & Korsmeyer, S.J. (1993). Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74, 609-619.
Pan, G., ORourke, K & Dixit, V.M. (1998). Caspase-9, Bcl-XL and Apaf-1 form a ternar complex. J. Biol. Chem. 273, 5841-5845.
Parrizas, M., Saltiel, A.R. & LeRoith, D. (1997). Insuline-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways. J. Biol. Chem. 272, 154-161.
Parsadanian, A.S., Cheng, Y., Keller.Peck C.R., Holtzman, D.M. ans Snider, W.D. (1998). Bcl-XL is an antiapoptotic regulator for postnatal CNS neurons. J. Neurosci. 18, 1009-1019.
Pettmann, B. & Henderson, C.E. (1998). Neuronal cell death. Neuron 20, 633-647.
Piñon, L.G.P., Middleton, G., & Davies, A.M. (1997). Bcl-2 is requiered for cranials sensory neuron survival at defined stages of embryonic development. Development 124, 4173-4178.
Rudel, T., & Bokoch, G.M. (1997). Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science 276, 1571-1574.
Salvesen, G.S & Dixit, V.M. (1997). Caspases: intracellular signaling by proteolysis. Cell 91, 443-446.
Shindler, K.S., Latham, C.B. & Roth, K.A. (1997). Bax deficiency prevents the increased cell death of immature neurons in bcl-X deficient mice. J. Neurosci. 17, 3112-3119.
Squier, M., Miller, A., Malknison, A. & Cohen, J. (1994). Calpain activation in apoptosis. J. Cell. Physiol. 159, 229-237.
Stennicke, H.R. & Salvesen, G.S. (1998). Properties of caspases. Biochem et Biophys Acta 1387, 17-31.
Tanaka, M., Sawada, M., Miura, M. & Marunouchi, T. (1998). Insulin-like growth factor-I analogue prevents apoptosis mediated through an interleukin-1b -like protease of cerebellar external granular later neurons: developmental stage-specific mechanism of neuronal cell death. Neuroscience 84, 89-100.
Thornberry, N.A., Bull, H.G., Calycay, J.R., Champan, K.T., Howard, A.D., Kostura, M.J., Miller, D.K.,Molineaux, S.M., Weidner, J.R., Aunins, J., Elliston, K.O., Ayala, J.M., Casano, F.J., Chin, J., Ding, J.F., Egger, L.A., Gaffney, E.P., Limjuco, G., Palyha, O.C., Raju, S.M., Rolando, A.M., Salley, J.P., Yamin, Y., Lee, T.D., Shively, J.E., MacCross, M.M., Mumford, R.A., Schmidt, J.A. & Tocci, M.J. (1992). A novel heterodimeric cysteine protease is required for interleukin-1b processing in monocytes. Nature 356, 768-774.
Thornberry, N.A. & Lazebnik, Y. (1998) Caspases: enemies within. Science 281, 1312-1316.
Troy, C.M., Stefanis, L., Prochiantz, A., Greene, L.A. & Shelanski, M.L. (1996). The contrasting roles of ICE family proteases and interleukin 1b in apoptosis induced by trophic factor withdrawal and by cooper/zinc superoxide dismutase downregulation. Proc. Natl Acad. Sci.USA 93, 5635-5640.
Vaux, D.L., Weissman, I.L. & Kim, S.K. (1992). Prevention of programmed cell death in Caenorhabditis elegans by human bcl-2. Science 258, 1955-1957.
Xue, D. & Horvitz, H.R. (1995). Inhibition of the Caenorhabditis elegans cell-death protease CED-3 by a CED-3 cleavage site in baculovirus p35 protein. Nature 377, 248-351.
Yao, R. & Cooper, G.M. (1995). Requirement for phosphatydilinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 267, 2003-2006.
Yuan, J.Y., Shaham, S., Ledoux, S., Ellis, M.H. & Horvitz, H.R. (1993). The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1b converting enzime.Cell 75, 641-652.
Zha, J., Harada, H., Yang, E., Jockel, J. & Korsmeyer, S.J. (1996). Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14-3-3 not Bcl-XL. Cell 87, 619-628.
Zou, H., Henzel, W.J., Liu, X., Lutschg, A. & Wang, X. (1997). Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90, 405-413.
Punteros de Interés.
Página de la biblioteca virtual dedicada a la apoptosis. Definiciones y artículos divulgativos (en inglés).
Página de la joven y muy activa "cell death society" con multitud de información sobre congresos y reuniones (en inglés).
Se pretender construir una página divulgativa en español.
